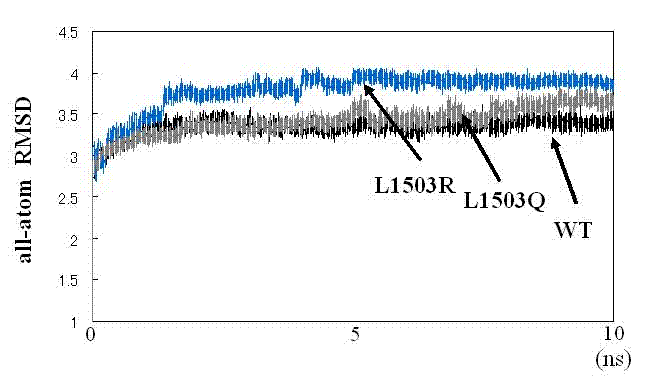
ダイナミクス計算→RMSD値
von Willebrand factor (VWF) A2の例で1503番目のロイシン(L)がアルギニン(R)またはグルタミン(Q)に変異した場合の野生型との蛋白構造の比較
T.Kashiwagi et.al Haemophilia (2008), 14, 556?563
10nsのダイナミクス計算
2.リガンドがそのエリアに結合できるか(結合に影響がある度合)
ligand-docking
3.変異により、構造が維持できないことの証明
1.リガンドドッキングサイト
2.不活性・活性構造に変化できなくなる
とにかく変異体モデルをつくってみたい
既に構造が解析されている場合、構造データより、PDBファイルの取得する。 まだ解析されていない場合、構造予測より、ホモロジーモデリングする。
PDBファイルの最適化
1. MOEを起動する。
2. File/Openで開く。
PDB fileの場合、ファイルを選択後、右のForce TypeをPDBにしてOKを押す。その後は2度OKを押す。
3. ウィンドウ右上ボタンの2つめ、Sequence Editorを開く。
4. Sequence EditorウィンドウのChainの項目が1つでない場合、H2O、リガンド等のchainを押し、Edit/Delete Selected Chainで削除する。
5. Select/Element/Heavy Atomを選ぶ。
6. Edit/Fixを選ぶ。
7. Compute/Energy Minimizeを選ぶ。左上に数字が現れ、しばらくすると終了する。
8.File/Saveでファイル名を指定して保存する。ファイル属性を.moeとしておくとわかりやすい。
(後で使うのでポテンシャルエネルギーを記録する。)
構造ファイルのチェック
1. Sequence EditorでMeasure/Protein Reportを選ぶ。
2. Protein Reportウィンドウ右上Outliers Onlyのチェックを入れる。
3. Protein Reportウィンドウ左下のCreateを押す。
4. Dihedrals, Bond Angles, Bond Lengths, Contactsをそれぞれクリックし、表示されるものをみる。
5. Protein Reportウィンドウ下のSaveボタンを押し、名前を指定して保存する。
6. Wordpadで保存したファイルを開き、プリントしておく。
変異モデル
1. Sequence Editorで、変異したいアミノ酸を選択する。
2. Edit/Rotamer Explorerを選ぶ。
3. Rotamer Explorerウィンドウ左下のGet from SEボタンを押す。
4. Rotamer Explorerウィンドウの上のアミノ酸を変異したいアミノ酸に変更してその右のExploreを押す。
5. Rotamer Explorerウィンドウ下中央のMutateを押す。
検討の仕方
1.構造ファイルのチェック
2.ポテンシャルエネルギーのチェック。野生型と比較。
3.変異モデルと元の構造で違いがあれば、そこを検討すればよい。
Last update 2017/08/25